Kotische, то, что ты предлагаешь, называют пастеризацией спирта. Но для неё нужна сотня тарелок, не меньше. Этого достаточно тяжело достичь в домашних условиях. Наверное есть смысл проводить эпюрацию сырца по принципу непрерывной ректификации для предварительного отсечения головных и промежуточных фракций, а затем обычной ректификацией эпюрата отсечь хвосты и концевые фракции. Я планировал заняться этим, но известные тебе обстоятельства пока меня тормознули.
Rudy
Академик
Питер
5.8K 1K
Отв.41 29 Июля 08, 07:24
Так внимательно посмотри, что Яровенко подразумевает под "накоплением"! Он под этим подразумевает повышенную концентрацию примеси, а вовсе не то, что она там накапливается!Rudy, 29 Июля 08, 05:00
Если на каком-то уровне концентрация повышается, значит имеет место накопление. Именно накопление. А если устроить отбор из середины колонны, мы накопленное там отведём и устраним это накопление.
...это не означает, что сама примесь накапливается в этой точке колонны! Она всегда пролетает колонну насквозь, если не реагирует с насадкой.
Не буду снова вставлять цитату из Яровенко. Ты всё равно их не читаешь. А напрасно. Яровенко очень доступно показывает почему примесь не может "улететь" Игорь, 29 Июля 08, 05:26
Ну, если так, то я могу тебе предложить идеальный способ удаления примесей . Перекрываем отбор, ждем, пока вся примесь не "накопится" в определенной точке колонны. Затем этот участок колонны перекрываем и сливаем. Затем продолжаем перегонку. ПОБЕДА! Вся примесь ушла из бака.
Kotische
Академик
Саратов
8.1K 2.5K
Отв.42 29 Июля 08, 23:45
то, что ты предлагаешь, называют пастеризацией спирта. Но для неё нужна сотня тарелок, не меньше.Игорь, 29 Июля 08, 05:52
Честно сказать не совсем понимаю, чем случай пастеризации тяжелее случая обычной ректификации. Имхо, если разделительная способность колонны будет плохая, то результат будет плачевным и в том и в другом случае... Разъебисни пожалуйста, в чем, в твоем понимании, заключается выигрыш "эпюрации сырца по принципу непрерывной ректификации" по сравнению с этой твоей "пастеризацией", с точки зрения унутриколониальных процессов...
Игорь
Академик
-
7.4K 3.7K
Отв.43 30 Июля 08, 04:41
я могу тебе предложить идеальный способ удаления примесей . Перекрываем отбор, ждем, пока вся примесь не "накопится" в определенной точке колонны. Затем этот участок колонны перекрываем и сливаем. Затем продолжаем перегонку. ПОБЕДА! Вся примесь ушла из бака. Rudy, 29 Июля 08, 07:24
То есть надо время от времени то небольшое количество флегмы и пара, которое имеется в данный момент в колонне, полностью вывести оттуда. Я предполагал для этого устраивать отбор всей флегмы без ее возврата в насадку. При этом в колонне прекратится движение веществ вниз, и через некоторое время мы получим во всей насадке и в дефлегматоре чистый кубовый пар, а температура пара в дефлегматоре сравняется (или почти сравняется) с температурой пара в кубе. Это равенство (или почти равенство) температур можно использовать для определения момента окончания "продувки". Естественно, после такой операции надо будет снова вводить колонну в режим, но это уже несложно. Навскидку 10-15 минут работы на себя должно быть достаточно. Итак, мой способ можно назвать продувкой путём прогрева колонны или продувкой паром. Rudy, то что предлагаешь ты, можно назвать промывкой путем охладжения колонны, или промывкой флегмой. То есть нужно перекрыть кран на входе в колонну, дать всему пару в колонне превратиться во флегму и стечь вниз, а затем открыть краник на небольшом боковом ответвлении в самом низу и слить скопившуюся флегму. Для того, чтобы смыть с насадки остатки, можно в дефлегматор влить некоторое количество спирта... или просто воды. Надо пробовать. Нельзя забывать о том, что при отключении колонны куб останется без связи с атмосферой, так что на этот момент нужно предусмотреть подключение обратного холодильника дабы не рвануло куб. Да, нагрев можно отключить, но если это сделать до того, как кран на колонну закрыт, вся накопившаяся гадость проскочит в куб и вывести из процесса её не удастся.
Не делая серьёзного анализа, на первый взгляд оба варианта - и продувка, и промывка должны быть эффективными. Промывка приведет к малым потерям продукта, и это плюс, но потребует некоторого усложнения системы, а это минус. В случае продувки плюсы и минусы меняются местами. Никакого усложнения "железа", но зато большее количество продукта попадет на вторичную переработку.
Предстоят эксперименты. Нужно определить эффективность промывки-продувки. И если эффект будет, нужно выяснить когда именно и как часто продувать-промывать. Это будет достаточно индивидуальное решение, зависящее от удерживающей способности колонны, состава и содержания примесей в сырце.
Rudy
Академик
Питер
5.8K 1K
Отв.44 30 Июля 08, 04:54, через 14 мин
То есть надо время от времени то небольшое количество флегмы и пара, которое имеется в данный момент в колонне, полностью вывести оттуда. Игорь, 30 Июля 08, 04:41
Да зачем же. У тебя же примеси "накапливаются" (т.е. их количество непрерывно растет) в колонне. Вот и подождем, когда в колонне "накопится" вся примесь из бака, а тогда и проделаем описанную операцию.
Игорь
Академик
-
7.4K 3.7K
Отв.45 30 Июля 08, 05:08, через 14 мин
Разъебисни пожалуйста, в чем, в твоем понимании, заключается выигрыш "эпюрации сырца по принципу непрерывной ректификации" по сравнению с этой твоей "пастеризацией", с точки зрения унутриколониальных процессов... Kotische, 29 Июля 08, 23:45
Вся суть в том, что очень эффективно работающая при ректификации колонна, при пастеризации теряет свою разделительную способность. Я сам не до конца разобрался почему, но попытаюсь объяснить тебе, может и сам пойму. :):) Давай для начала рассмотрим тарельчатую колонну. В кубе 96,4%, в дефлегматоре 96,4%, на каждой тарелке 96,4%. Пар с содержанием спирта 96,4%, барботируя через флегму 96,4% на нижней тарелке, даже не заметит её. Нет перепада температур - нет конденсации и испарения - нет эффекта, нет изменения состава флегмы и пара. То есть физическая тарелка есть, а теоретической ступени (единицы переноса) нет. Значит КПД нижней тарелки равно нулю. Теперь этот пар, не претерпев никаких изменений, барботирует через флегму второй тарелки. А там тоже 96,4%. Результат в точности такой же, как и в первом случае. . . . Теперь этот пар, не претерпев никаких изменений, барботирует через флегму 100-ой тарелки. А там тоже 96,4%. Результат в точности такой же, как и в первых 99-ти случаях.
Допустим, что я всё утрировал процентов на 5. В таком случае КПД колонны составляет 5%, и колонна, разделяющая при ректификации как 100-тарелочная, при пастеризации проявляет себя всего на 5ТТ.
Про эпюрацию обязательно продолжу. Спокойной ночи.
Переходим к насадочной колонне. Поднимающийся 96,4%-ный пар, встречая на своём пути опускающуюся 96,4%-ную флегму, не заметит её присутствия, не станет с ней тепломассообмениваться, в неизменном виде пройдет в дефлегматор, и в том де виде попадёт в куб.
Игорь
Академик
-
7.4K 3.7K
Отв.46 30 Июля 08, 05:13, через 5 мин
Да зачем же. У тебя же примеси "накапливаются" (т.е. их количество непрерывно растет) в колонне. Вот и подождем, когда в колонне "накопится" вся примесь из бака, а тогда и проделаем описанную операцию.
Концентрация примесей в паре-флегме растет, и через некоторое время, обусловленное удерживающей способностью колонны, они либо смываются флегмой в куб, либо частично выносятся паром в дефлегматор, загрязняя продукт. Причем этот процесс не имеет характера прагматехниковской ступеньки, а достаточно долгое время сопровождает выход чистого продукта. Поэтому и открыта эта ветка.
P-Alex
Научный сотрудник
пгт.Палех
1.2K 175
Отв.47 30 Июля 08, 08:40
Предстоят эксперименты. Нужно определить эффективность промывки-продувки. И если эффект будет, нужно выяснить когда именно и как часто продувать-промывать. Это будет достаточно индивидуальное решение, зависящее от удерживающей способности колонны, состава и содержания примесей в сырце. Игорь, 30 Июля 08, 04:41
Интересная тема... с нетерпением жду продолжения
Rudy
Академик
Питер
5.8K 1K
Отв.48 30 Июля 08, 09:11, через 31 мин
Да зачем же. У тебя же примеси "накапливаются" (т.е. их количество непрерывно растет) в колонне. Вот и подождем, когда в колонне "накопится" вся примесь из бака, а тогда и проделаем описанную операцию.
Концентрация примесей в паре-флегме растет, и через некоторое время, обусловленное удерживающей способностью колонны, они либо смываются флегмой в куб, либо частично выносятся паром в дефлегматор, загрязняя продукт.Игорь, 30 Июля 08, 05:13
А вот это совершенно справедливо. Наконец-то ты понял, что никакого "накопления" примесей в колонне не может быть.
Причем этот процесс не имеет характера прагматехниковской ступеньки, а достаточно долгое время сопровождает выход чистого продукта. Поэтому и открыта эта ветка.Игорь, 30 Июля 08, 05:13
И это в значительной степени справедливо. Только ты не совсем правильно ругаешь прагматехников, их график описывает совершенно иной процесс. И он, в целом, верен. Но на приведенном ими графике совершенно не заметны мелкие подробности, которые имеют величину порядка десятых и сотых долей процента, которые и портят тебе жизнь. У них начальное количество - порядка 100 грамм. А сколько метилового спирта нужно, чтобы испортить продукт? На их графике такие количества просто не видны. Так что не ругай их зря, обычное дело. Просто каждая ступенька на их графике имеет экспоненциальные (а, на самом деле совсем не экспоненциальные) хвосты, которые по величине невелики, но тянутся далеко и заходят на соседние ступеньки, что и вызывает рассматриваемые проблемы.
Раз уж диалог пошел конструктивный, добавлю "загадочную" фразу: "Количество примеси метилового спирта прямо пропорционально (примерно) мощности нагревателя". Или, иными словами, количество легких (в смысле легколетучих) примесей можно существенно снизить уменьшив скорость ректификации. Причем зависимость, похоже, пропорциональная. Чтобы обосновать это заявление, потребуется очень долгое разбирательство и я не знаю, стоит ли даже начинать, тем более, что и сам еще не все до конца понимаю. А практически, это будет звучать примерно так. В начале отгонки головы, следует установить большое флегмовое число и держать температуру пара в дефлегматоре чуть ниже 78 градусов, регулируя мощность нагревателя. Она должна быть минимально возможной. Чем дольше продержать колонну в таком режиме, тем меньше будет легких примесей в спирте. Потом - обычный режим.
Игорь
Академик
-
7.4K 3.7K
Отв.49 31 Июля 08, 17:58
Rudy, метиловый спирт легколетуч только в чистом виде. Когда эта гадость присутствует в виде небольшой примеси в водно-спиртовом растворе, она совсем даже не легколетуча. Её относят не к головным и даже не к хвостовым, а к концевым примесям. Сейчас я не имею под руками графиков коэффициентов ректификации, но если ты посмотришь на табличку, заметишь, что при кубовых концентрациях этилового спирта, с котрых мы начинаем ректификацию, метиловый спирт испаряется хуже, чем этиловый, и его содердание в парах будет ниже, чем в растворе, в отличие от этилового, концентрация которого в парах почти всегда (до азеотропной точки) больше, чем в испаряющейся жидкости. Но метиловый спирт - экзотика. При испольовании сахара для приготовления браги он практически не образуется, а при кубовых концентрациях спирта в браге 12-16% он настолько трудноиспарим, что его концентрация в парах, а значит и в самогоне, в разы ниже, чем в браге. ------------------------------------ Я пробовал делать длительный отбор голов при огромных флегмовых числах и малой мощности нагрева. Именно неудача этого способа заставила меня лезть в дебри промежуточных фракций и искать причину неприятностей в их влиянии.
Началось с того, что при ректификации 20 литров 40%-ного самогона я получил 500 мл неисправимых голов, 2 литра спирта с лёгким запахом примесей и чуть больше 5 литров нормального спирта. Хвостов не отбирал, они замечательно отсекаются при моём старт-стоповом способе управления отбором по температуре.
Я решил, что у меня "размазались головы" и решил тщательно отделить их, поэтому вторые 20 литров того же самогона подверг "медленному головоотделению". При мощности нагревателя порядка 400 ватт я всю ночь держал капельный отбор. За 12 часов отобрал чуть меньше 600 мл , после чего постепенно увеличил мощность до 700 ватт и установил отбор 500 мл/час. Общее количество спирта в кубе было около 8 литров - такое же, как и при первой - обычной - ректификации.
Результаты меня поразили. Количество спирта, пованивающего эфирно-альдегидной фракцией, составило 4 литра, нормального спирта - чуть более трёх. Фиаско.
Причина этого провала на мой взгляд может заключаться в том, что при длительном кипячении и многократном испарении-конденсации на насадке происходят каталитические реакции, в результате которых образуются дополнительные порции примесей. Тут стоит подумать о целесообразности установок периодического действия с большим кубом и колонной низкой производительности. В этом плане идеальна колонна непрерывного действия, где время обработки единицы объема сырца минимально. Я уже подумываю о создании автоматизированной колонны непрерывного действия с производительностью 30 мл/час с рекуперцией тепла и воздушным охлаждением. За неделю она выдаст литров 5 спирта, значит мне нужно будет еженедельно заливать в её питающий бак 12-13 литров 40%-ного самогона, кторые мжно получить из 10 кг сахара. Если питающий бак сделать литров на 50, можно не корячиться с еженедельной перегонкой браги, а делать это раз в месяц. Потребляемая мощность такой установки с учетом вентилятора охлаждения не превысит 150 - 200 ватт, а размеры колонны могут быть действительно игрушечными. Но это для меня пока отдаленная перспектива, нужно сперва хорошо понять происходящие процессы. Опираясь на такую установку можно подумать и о топливном этаноле.
Rudy
Академик
Питер
5.8K 1K
Отв.50 31 Июля 08, 18:14, через 17 мин
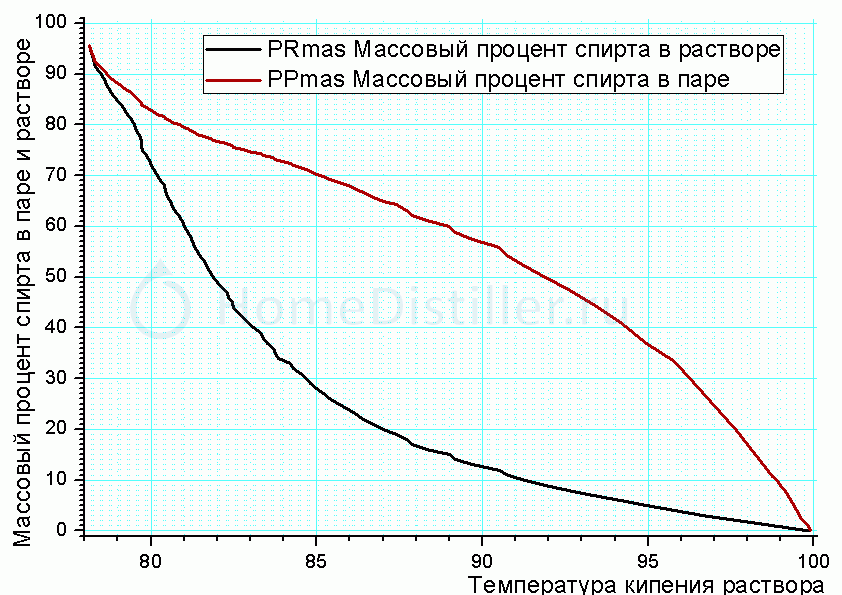
Ух, какую путаницу ты устроил. Я думаю, ты и себя уже запутал. Если действительно хочешь разобраться, давай о-о-очень медленно и маленькими кусочками. Ты знаешь диаграмму концентрации раствор-пар для этилового спирта как функции температуры кипения водного раствора? А теперь найди такую же для метилового спирта и нарисуй их вместе на одном графике (если не лениво, скинь этот график в форум). И посмотри, что будет происходить при медленном нагреве раствора, содержащего равные доли (допустим по 5% масс) этилового и метилового спиртов. Это будет то, что ты получишь при простой перегонке без ректификации. А потом разберемся и с ней. Данные по этиловому спирту у меня есть. Если найдешь данные по метиловому - просто скинь мне ссылку или таблицу, графики я сделаю. Примерно в таком виде:
SpirtConPar_Rastv_Tkip.gif Промежуточные и концевые фракции. . Очистка спирта.
Игорь
Академик
-
7.4K 3.7K
Отв.51 01 Авг. 08, 08:57
Ух, какую путаницу ты устроил. Я думаю, ты и себя уже запутал Rudy, 31 Июля 08, 18:14
Нет, эту путаницу внесла Прагма, подменив понятие летучести примеси температурой её кипения.
...содержащего равные доли (допустим по 5% масс) этилового и метилового спиртов.... Это будет то, что ты получишь при простой перегонке без ректификации...
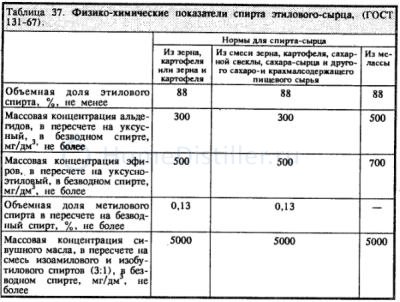
Rudy, а это уже проявления твоего недопонимания. Зачем тебе изучать ситуацию, которая никогда на практике не встречается? Прикинь. Содержание метанола в самом хреновом спирте-сырце не превышает 0,15% (Яровенко, табл.34 стр.320). Это для 80%-ного сырца. В нашем 40%-ном его соответственно может быть не больше 0,08%. К концу ректификации, когда весь этанол покидает куб, объем куба уменьшается на 40%. Если весь метанол к тому времени останется в кубе, его концентрация всё равно не превысит 0,2%. Так что вариант "5% того и 5% того" из области фантастики. Во всей области ректификации концентрация метанола настолько мала, что на его испаряемость влияют не межмолекулярные связи метанола, определяющие его температуру кипения, а связи между молекулами метанола с молекулами основных компонентов смеси. Построить график несложно. Исходные данные - начальная концентрация метанола (я возьму 0,2%), твои графики по 10-тарельчатой колонне и кривая 23 на рис.93 стр.323 у Яровенко. Промежуточные и концевые фракции. . Очистка спирта. Таким образом я смогу проследить концентрацию метанола на каждой тарелке при различных концентрациях кубового раствора. К обеду сделаю.
Rudy
Академик
Питер
5.8K 1K
Отв.52 01 Авг. 08, 09:36, через 40 мин
Для начала законный, чисто риторический, вопрос. Если примеси так мало, стоит ли с ней столь тяжко бороться? Понятно, что твой нос и вкус скажут обратное, но все же. Она, возможно, противна для вкуса, но не смертельна. А ты людей пугаешь.
Какая разница какой процент. Я хотел чтобы ты посмотрел эти кривые для того, чтобы лучше понимать, что и как происходит. А ты опять попытался перескочить через несколько ступенек и промахнулся. Так считать нельзя.
Давай я опять скажу "загадочную фразу": "Если в паре из бака процент метанола превышает некоторую величину, зависящую от параметров колонны (сотые или десятые доли процента, или проценты, в зависимости от качества колонны и режима ее работы), то циркуляция паров метанола в верхней части колонны не пропустит этанол на выход колонны. При этом, температура пара на выходе колонны будет соответствовать температуре кипения метанола". Иными словами, пока процент метанола в баке не опустится ниже некоторой величины, на выходе колонны вообще (практически) не будет этанола. Попробуй покажи мне эту ситуацию на своих графиках из Яровенко. А вот на кривой прагматехников это очевидно. Ты просто не очень хорошо представляешь себе как работает ректификационная колонна. То, что я говорил ранее о режиме с минимальной мощностью, это способ заставить колонну вести себя так, как написано в "загадочной фразе" предельно долго, т.е. уменьшить концентарацию метанола, при которой этанол не пролезает на выход. При этом, естественно, сам метанол постепенно уходит из бака. Именно этот процесс и определит тот начальный процент метанола, который потом полезет в этанол. Чем лучше мы выберем из бака метанол в этом режиме, тем меньше его будет в конечном продукте.
Про кривые из Яровенко - еще раз повторю, что пользоваться ими можно только точно зная как и в каких условиях они получены. А учебник Яровенко не только не объясняет этого, но даже ссылок не дает. А те ссылки, которые есть в Дороше недоступны.
Игорь
Академик
-
7.4K 3.7K
Отв.53 01 Авг. 08, 10:43
Для начала законный, чисто риторический, вопрос. Если примеси так мало, стоит ли с ней столь тяжко бороться?
Ответ прост. Здесь мoжно посмотреть гостовскую концентрацию примесей в сырце. Суммируя получаем, что общее количество примесей в спирте-сырце в переводе на безводный спирт нахдится в пределах 0,5 - 1%. Вот с этими полпроцентами и боремся. Кто не желает бороться - пьет безвредный самогон, кто-то борется с тысячными долями процента. У каждого свой путь.
Содержание некоторых примесей в тысячные доли процента делает спирт несоответствующим стандарту. Примесь некоторых веществ, не определяемая физическими или химическими методами, определяется только органолептически и может сделать спирт весьма противным.
Rudy, не обижайся, но у меня не так много свободного времени, чтобы -надцатый раз объяснять одно и то же. Я иду дальше, а ты (если есть желание) догоняй. Если данные Яровенко противоречат прагмовской информации, которая для тебя удобна, можешь её не использовать, твоё дело.
Игорь
Академик
-
7.4K 3.7K
Отв.54 01 Авг. 08, 11:05, через 23 мин
Я пошел. Для начала основные определения.
Коэффициент испаряемости вещества показывает отношение его концентрации в парах к концентрации в испаряющейся жидкости. В применении к нашему случаю этот коэффициент показывает во сколько спирт испаряется лучше воды при данной спиртуозности исходного раствора.
Коэффициент ректификации примеси показывает отношение коэффициента испаряемости примеси к коэффициенту испаряемости этанола в условиях, когда этанол и вода являются основными компонентами смеси, а содержание примеси в растворе незначительно.
Коэффициент испаряемости нам знаком. При нагревании раствора этанола в воде происходит испарение и воды и этанола. Но этанол в данных условиях испаряется в N раз лучше воды, поэтому его содержание в парах будет в N раз выше, чем было до первого испарения. Число N и есть коэффициент испарения спирта.
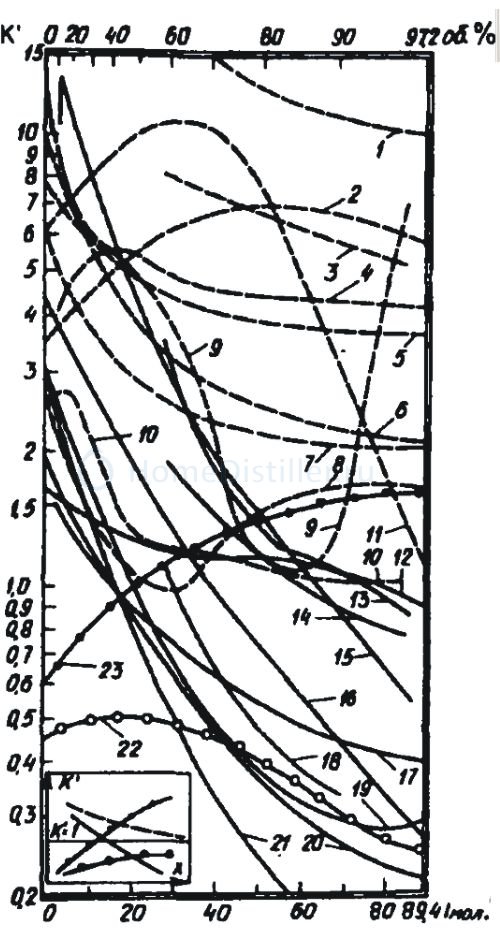
Коэффициент ректификации примеси пока знаком меньше. Для разных примесей он приведен в страшном графике. Для удобства я выдернул из этого графика метанол и более-менее причесал его.
П Р О Д О Л Ж А Ю
Сергей Юрьич из ТАГАНРОГА
Новичок
ТАГАНРОГ
2
Отв.55 02 Авг. 08, 22:44
Позволю себе поделиться с коллегами тем, что нарыл и до чего допёр. Итак, Прагма со своей инструкцией ввела нас в заблуждение. К моему огромному сожалению, примеси не идут одна за другой в узел отбора в соответствии с температурой их кипения. Они ведут себя по другому. Степень их испаряемости определяется коэффициентом ректификации. Если этот коэффициент больше единицы, значит в парах содержание примеси будет больше, чем в жидкости, и наоборот. Если коэффициент испаряемости меньше единицы, то при испарении примесь будет задерживаться во флегме, не особенно стремясь испариться. Весь фокус заключается в том, что коэффициент ректификации примеси не константа. Он изменяется при изменении концентрации спирта, в которой эта примесь сидит. График коэффициентов ректификации разных примесей на первый взгляд напоминает на макаронной фабрике. Но в этих кривульках заложена измена. Например, в нашей браге содержался в некотором количестве бутанол. При перегонке браги её крепость менялась - скажем - от 12% до нуля. В этом диапазоне спиртуозности коэффициент ректификации (КР) бутанола примерно равен трём. Это значит, что концентрация бутанола в парах (а значит и в самогоне) будет втрое больше, чем была в браге. За время перегонки весь бутанол покинет брагу и накопится в самогоне. Теперь этот самогон с увеличенным содержанием бутанола мы будем ректифицировать. Ректификация начинается из сырца с крепостью 40%, затем пар проходит всю насадку, проходя через все концентрации спирта вплоть до 96-97%. Давайте посмотрим как поведет себя бутанол. При 40% он имеет коэффициент ректификации (КР) 1,5, то есть его концентрация в парах становится больше, чем была в сырце. По мере обеднения сырца к-нт ректификации бутанола растёт, значит он весь испарится и попадёт в колонну. Какая его дальнейная судьба? Попав в зону насадки, где концентрация спирта примерно равна 60%, КР бутанола становится равным 0,5. То есть его концентрация в парах становится вдвое меньше, чем была в жидкости. И чем выше по градусам (и по колонне), тем хуже он испаряется. В куб вместе с флегмой бутанол вернуться не может, так как ниже по колонне спиртуозность ниже, а КР бутанола больше. Он испаряется , поднимается немного выше и сноваа конденсируется. В результате он насышает какую-то область колонны. Каждая промежуточная примесь оккупирует некоторую область колонны. Так, например, изомасляноэитловый эфир накапливается в зоне насадки, где спиртуозность равна 90%, изовалерианоэтиловый - 85%, уксусноизоамиловый эфир - 68%, изовалерианоизоамиловый эфир - 60%. Список можно продолжить. Но не в его длине смысл. Главный вопрос в том, куда деваются эти примеси. Пути вниз им нет, там каждую секунду падает спиртуозность, а значит повышается испаряемость этих примесей. Да и колонна не резиновая. Рано или поздно она переполнится. Значит волей-неволей эти примеси будут вытеснены в дефлегматор и благополучно нами выпиты. А не хотелось бы. Давайте искать пути борьбы с этими граблями. Игорь, 24 Июня 08, 20:12
Предлагаю!Вместо колонны непрерывного действия( с дырками в разных местах). При перегонке через обычную нашу колонну НЕ СЛИВАТЬ В ОДНУ банку всю"полезную"фракцию(сместе с бутанолом и прочей дрянью),а последовательно делить дистиллят на три-пять,(а можно и лучше больше) разных банок.Таким образом в каждой из банок соберётся смесь эт.спирта и какой ни будь(одной!!,а не всех сразу!!) гадости.Ну а затем,разбавление водой и повторная перегонка,с отсечением как обычно головы и хвоста...Правда придётся "первичную" перегонку проводить несколько порций самогона,что б потом слить вместе банки с одинаковыми номерами(и примесями),и получить количество "достойное" для повторной перегонки...Как идейка?
Kotische
Академик
Саратов
8.1K 2.5K
Отв.56 03 Авг. 08, 01:07
а затем,разбавление водой и повторная перегонкаСергей Юрьич из ТАГАНРОГА, 02 Авг. 08, 22:44
При следующей перегонке горячая вода опять выдавит все примеси в целевую фракцию, итого - результат = 0!
А когда на колонне имеется значительный температурный градиент то получить хорошее разделение, имхо, абсолютно не реально. Промежуточные и концевые фракции. . Очистка спирта.http://absintheclub.ru/read/viewtopic.php?p=30529#30529
Сергей Юрьич из ТАГАНРОГА
Новичок
ТАГАНРОГ
2
Отв.57 03 Авг. 08, 07:57
а затем,разбавление водой и повторная перегонкаСергей Юрьич из ТАГАНРОГА, 02 Авг. 08, 22:44
При следующей перегонке горячая вода опять выдавит все примеси в целевую фракцию, итого - результат = 0!
А когда на колонне имеется значительный температурный градиент то получить хорошее разделение, имхо, абсолютно не реально. Промежуточные и концевые фракции. . Очистка спирта.http://absintheclub.ru/read/viewtopic.php?p=30529#30529
Kotische, 03 Авг. 08, 01:07
Ну так давайте второй раз не разбавлять..или совсем чуть-чуть..
P-Alex
Научный сотрудник
пгт.Палех
1.2K 175
Отв.58 15 Окт. 08, 16:50
А что если для вывода промежуточных фракций использовать схему Селянина (которая с поварешкой) для отвода стекающей в куб флегмы... но с одним добавлением: в определенном месте по высоте колонны мы вводим воду... соответственно промежуточные фракции из за уменьшения крепости будут желать убежать в куб, но попадут в поварешку и будут изгнаны... плюс еще у отводимой флегмы будет крепость пониже
Klim
Научный сотрудник
Новокузнецк
1.7K 542
Отв.59 15 Окт. 08, 17:09, через 19 мин
Все разговоры о фракциях на уровне теорий,ну кроме самых-самых.Нам бы мужика с газо-хромо-спектральным(или как там его) анализом.Чтоб поэтапно все фракции проанализировать.Прям сейчас меня больше волнует,что прокладка(якобы силикон)даёт столько вони при прокрутки колонны на воде,что все примеси меркнут.



 :):)
:):)




{kind=link}